 Catalysts and batteries
Catalysts and batteries
Establishment of Molecular Technology Toward the Creation of New Functions (JST CREST)
Development of Asymmetric C-H Bond Activation Catalysts Guided by Quantum Simulations (JST ACT-C)
-

- Computation

-

- Information

- Developing the AFIR method for performing automated searches of chemical reaction paths
- Using a model function to add artificial force to reactant pairs in the AFIR method
- Enhancing generalization by combining various existing computational techniques
Predicting reaction products and paths automatically
The discovery of innovative chemical reactions can produce a profound social and economic ripple effect. However, at the present time new reactions can only be discovered through trial and error involving innumerable experiments. Constructing a computational method that accepts such input as reactants and catalysts and automatically predicts products, byproducts, and the reaction paths by which they are produced based on quantum chemical calculations would drastically change the speed at which reactions and materials are developed. This is precisely the objective of our research.
Pushing a reactant pair together to eliminate the energy barrier
In traditional computational chemistry, chemical problems cannot be analyzed without reasonable estimation of the reaction mechanism (excluding extremely short reactions of about one hundred millionth of a second). As a consequence, mistakes in estimation yield incorrect results. An automated method for searching chemical reaction paths is needed to achieve systematic analysis and reliable prediction. The artificial force induced reaction (AFIR) method that I will introduce here was developed in 2010 and has since made great strides toward generalization.
The essence of the AFIR method is to push reactants together to force a reaction. For example, in the case of atoms A and B, a potential energy barrier is present between the two. When an artificial force is applied to both atoms to push them together, the barrier is eliminated, and A and B drop in a potential well, allowing for a quick investigation of the reaction mechanism. Specifically, the product is found using an AFIR function that adds the αrAB term (where α is a proportional constant), which is proportional to the distance rAB between A and B, to the potential energy and repeatedly performing an operation to minimize the AFIR function from its initial state. Further, the actual reaction path and energy are determined by setting the constant α for artificial force to 0, returning the atoms to their original potential, and repeating the calculation. With this method, it is now easy to find the positions of transition states.
The AFIR method is not only for use with intermolecular reaction, but can also be applied to reactions within a single molecule by pushing any of various fragments in the molecule together or pulling them apart. In this way, reaction paths can be systematically searched, beginning from the initially inputted structure, allowing for the discovery of numerous new local minima. Using each of the acquired local minima as the starting point and performing searches for all of the local minima, it is possible to search all possible reaction paths for the given chemical composition. We call the network of reaction paths acquired through these searches the “global reaction route map (GRRM).”
Combining various computational techniques to achieve generalization
Computational complexity becomes problematic for full searches when the number of atoms becomes large. Therefore, we introduced various options to perform a limited (partial) search so as to be applicable to reaction systems configured of a few tens of atoms up to about 100. For example, we have been making great strides in analyses on the mechanisms of organic synthesis reactions and the selectivity of products.
The range of applications continues to be expanded by combining the AFIR method with various computational techniques, such as multi-state algorithms for applying a search method used for photoreactions in which a plurality of potential energy surfaces take part in the reaction; a quantum mechanics/molecular mechanics (QM/MM) approach and a microiteration technique for treating reactions in huge systems like enzymatic reactions; periodic boundary conditions needed to handle crystals and other solids. At present the program supports organic synthesis reactions, metal cluster catalysis, solid surface reactions, photoreactions, enzymatic reactions, crystal phase transitions, and many other types of chemical reactions. Our future goal is to introduce acceleration specific to individual reaction systems of interest to be used for screening such systems prior to experimentation.
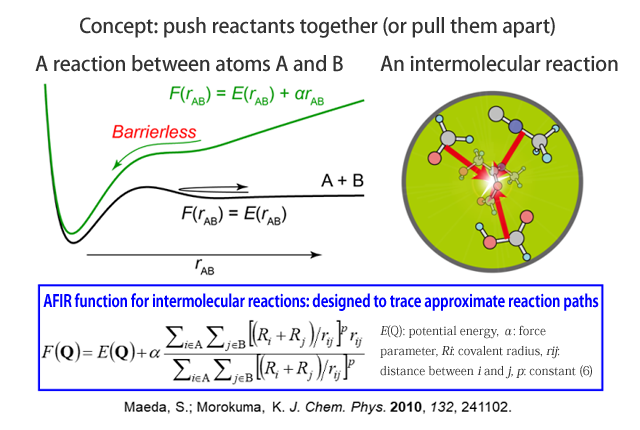
Figure 1 Artificial force induced reaction (AFIR) method
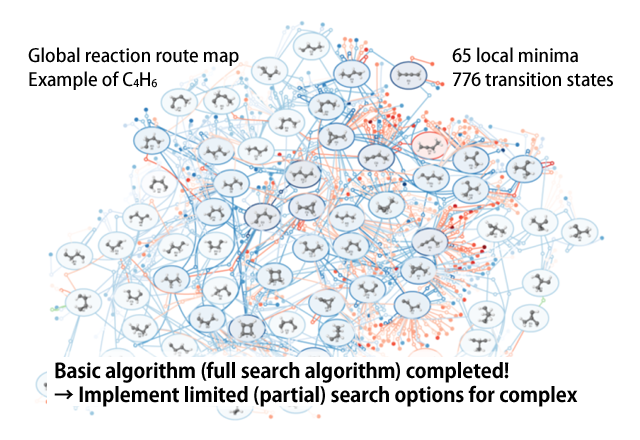
Figure 2 Global reaction route map (GRRM)

Satoshi Maeda
Theoretical Chemistry Lab.
Department of Chemistry, Faculty of Science
Hokkaido University
References:
- [1] S. Maeda, K. Morokuma, J. Chem. Phys. 2010, 132, 241102.
- [2] S. Maeda, Y. Harabuchi, M. Takagi, K. Saita, K. Suzuki, T. Ichino, Y. Sumiya, K. Sugiyama, Y. Ono, J. Comput. Chem. 2018, 39, 233.
- [3] S. Maeda, K. Sugiyama, Y. Sumiya, M. Takagi, K. Saita, Chem. Lett. 2018, 47, 396.